Chrome
Chrome Firefox
Firefox Opera
Opera Safari
Safari IE
IE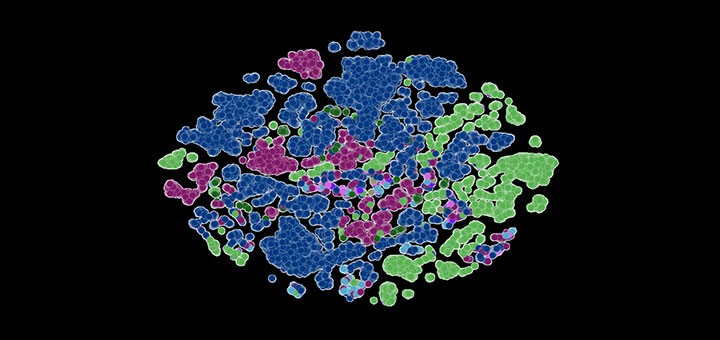
COVID-19 研究特別賞を受賞したアルゴンヌ国立研究所、NVIDIA、シカゴ大学などの研究者チームがゲノム スケールのデータを処理する最先端のモデルを開発
※本ブログは11月17日のゴードン ベル賞受賞者の発表に伴い、内容を更新しています
ハイ パフォーマンス コンピューティング ベースの COVID-19 研究に対するゴードン ベル特別賞の受賞チームが、大規模言語モデル (LLM) に、ゲノミクス、疫学、およびタンパク質工学における洞察を深めることが可能な遺伝子配列を教えました。
10 月に公開されたこの画期的な研究は、アルゴンヌ国立研究所、NVIDIA、シカゴ大学などの 20 人を超える学術研究者と商業研究者による共同研究です。
研究チームは、COVID-19 の背後にあるウイルスである SARS-CoV-2 で懸念される変異株を予測するために遺伝子変異を追跡し、LLM をトレーニングしました。これまでに生物学に適用されたほとんどの LLM は、低分子またはタンパク質のデータセットでトレーニングされていますが、このプロジェクトは生のヌクレオチド配列 (DNA と RNA の最小単位) でトレーニングされた最初のモデルの 1 つです。
「タンパク質レベルから遺伝子レベルのデータに移行することで、新型コロナ変異株を理解するためのより優れたモデルを構築できる可能性があるという仮説を立てました」と、プロジェクトを率いたアルゴンヌ国立研究所の計算生物学者、アービンド ラマナサン (Arvind Ramanathan) 氏は述べています。「ゲノム全体とその進化に現れるすべての変化を追跡するようにモデルをトレーニングすることで、新型コロナだけでなく、十分なゲノムデータがあれば、あらゆる病気についてより良い予測を行うことができます」
ハイ パフォーマンス コンピューティングのノーベル賞と見なされているゴードン ベル賞は、今週の SC22 カンファレンスにおいて、世界中 10 万 人以上のコンピューティングの専門家を代表する Association for Computing Machinery により授与されました。2020 年以降、同グループは、HPC を使用して新型コロナの理解を進める優れた研究に対して特別賞を授与しています。
4 文字言語での LLM のトレーニング
LLM は長い間、人間の言語でトレーニングされてきました。人間の言語は、通常、数十文字で構成され、数万の単語を並べ、より長い文や段落に結合されています。一方、生物学の言語では、ヌクレオチドを表す 4 文字しかなく、DNA では A、T、G、C、RNA では A、U、G、C が遺伝子として異なる配列に配置されています。
文字数が少ない方が AI にとっては簡単に思えるかもしれませんが、生物学の言語モデルは実際にははるかに複雑です。これは、ヒトでは 30 億個以上のヌクレオチド、コロナウイルスでは約 3 万個のヌクレオチドで構成されるゲノムを、明確で意味のある単位に分解するのが難しいためです。
「生命の暗号を理解する上で大きな課題となるのは、ゲノム内の配列決定情報が非常に膨大であることです」とラマナサン氏は説明します。「ヌクレオチド配列の意味は、次の文や段落よりもはるかに離れた別の配列の影響を受ける可能性があります。本の章に相当する文字数の距離に達することもあります」
本プロジェクトの NVIDIA の研究者は、LLM が約 1,500 ヌクレオチドの長い文字列を文のように処理できるようにする階層的な拡散方法を設計しました。
「標準言語モデルは、一貫した長いシーケンスを生成し、さまざまな変異株の根底にある分布を学習することが困難です」と語るのは、NVIDIA の AI 研究のシニア ディレクターであり、カリフォルニア工科大学のコンピューティング + 数理科学部門で名誉ある Bren 教授職の肩書を持つ、論文の共著者であるアニマ アナンドクマール (Anima Anandkumar) です。「我々はより詳細なレベルで動作する拡散モデルを開発しました。これにより、現実的な変異株を生成し、より良い統計を取得できます」
懸念される新型コロナ変異株の予測
チームは最初に、Bacterial and Viral Bioinformatics Resource Center (細菌やウイルス感染症の研究を支援するための情報システム) のオープンソース データを使用し、細菌のような単細胞生物である原核生物の 1 億 1000 万を超える遺伝子配列で LLM を事前トレーニングしました。次に、新型コロナウイルスの 150 万の高品質ゲノム配列を使用してモデルをファインチューニングしました。
より広範なデータセットで事前トレーニングすることにより、研究者はモデルが将来のプロジェクトで他の予測タスクに一般化できることも確認しました。このような機能を備えた最初の全ゲノムスケール モデルの 1 つになりました。
新型コロナのデータでファインチューニングすると、LLM はウイルスの変異株のゲノム配列を区別することができました。また、独自のヌクレオチド配列を生成し、新型コロナゲノムの潜在的な突然変異を予測することもできました。これは、科学者が懸念される将来の変異株を予測するのに役立ちます。
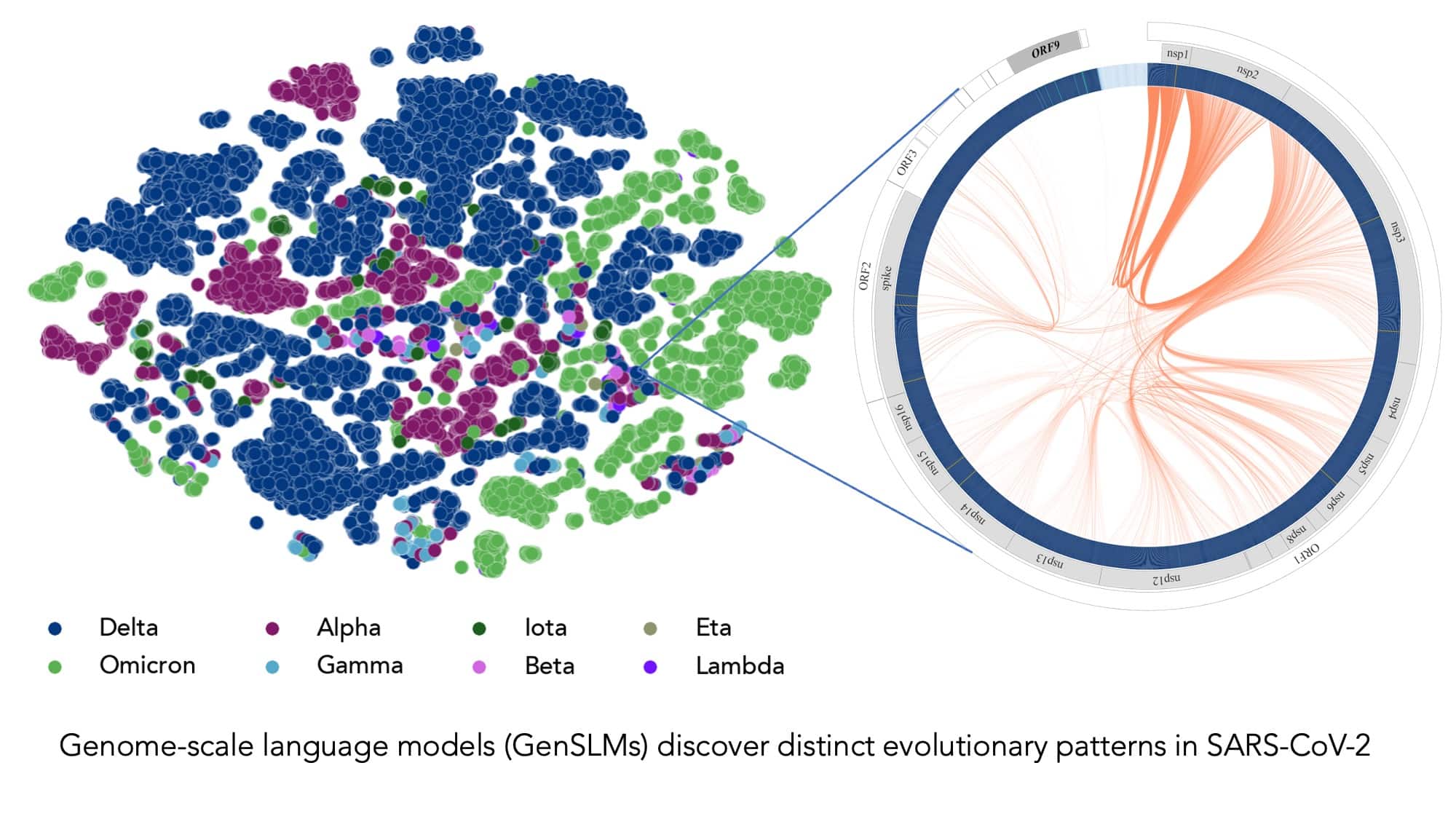 1年分のSARS-CoV-2ゲノムデータで学習させたモデルは、様々なウイルス株を区別して推論することができます。左の図は、配列が決定されたSARS-CoV-2ウイルスの株を、変異型ごとに色分けしたものです。右の図は、ある特定のウイルス株を拡大したもので、この株に特有のウイルスタンパク質間の進化的な結合を捉えています。
1年分のSARS-CoV-2ゲノムデータで学習させたモデルは、様々なウイルス株を区別して推論することができます。左の図は、配列が決定されたSARS-CoV-2ウイルスの株を、変異型ごとに色分けしたものです。右の図は、ある特定のウイルス株を拡大したもので、この株に特有のウイルスタンパク質間の進化的な結合を捉えています。画像提供:アルゴンヌ国立研究所のBharat Kale氏、Max Zvyagin氏、Michael E. Papka氏
「ほとんどの研究者は、新型コロナウイルスのスパイクタンパク質、特にヒト細胞と結合するドメインの変異を追跡してきました」とラマナサン氏は述べています。「しかし、ウイルスゲノムには、頻繁に突然変異を起こし、理解することが重要な他のタンパク質があります」
このモデルは、AlphaFold や OpenFold などの一般的なタンパク質構造予測モデルと統合することもでき、研究者がウイルス構造をシミュレートし、遺伝子変異がウイルスの宿主への感染能力にどのように影響するかを研究するのに役立つと論文は述べています。OpenFold は、デジタル生物学および化学アプリケーションに LLM を適用する開発者向けの NVIDIA BioNeMo LLM サービスに含まれる事前トレーニング済み言語モデルの 1 つです。
GPU アクセラレーテッド スーパーコンピューターによりAI トレーニングをスーパーチャージ
チームは、アルゴンヌ国立研究所 の Polaris、米国エネルギー省の Perlmutter、NVIDIA 社内の Selene システムを含む NVIDIA A100 Tensor コア GPU を搭載したスーパーコンピューターで AI モデルを開発しました。これらの強力なシステムにスケールアップすることで、トレーニングで 1,500 エクサフロップスを超えるパフォーマンスを達成し、これまでで最大の生物学的言語モデルを作成しました。
「我々は現在、最大 250 億のパラメーターを持つモデルを扱っており、将来的にはこれが大幅に増加すると予想しています」とラマナサン 氏は述べます。「モデルのサイズ、遺伝子配列の長さ、必要なトレーニング データの量は、何千もの GPU を備えたスーパーコンピューターによって提供される複雑な計算の必要性を示しています」
研究者たちは、25 億のパラメーターを持つモデルのバージョンをトレーニングするのに約 4,000 基のGPU で 1 か月以上かかったと推測します。すでに生物学の LLM を調査していたチームは、論文とコードを公開する前にプロジェクトに約 4 か月を費やしました。GitHub ページには、他の研究者が Polaris と Perlmutter でモデルを実行するための手順が含まれています。
GPU に最適化されたソフトウェアのハブである NVIDIA NGC でアーリー アクセス で利用可能な NVIDIA BioNeMo フレームワークは、複数の GPU にわたって大規模な生体分子言語モデルをスケーリングする研究者をサポートします。創薬ツールのコレクションである NVIDIA Clara Discovery の一部であるこのフレームワークは、化学、タンパク質、DNA、および RNA のデータ形式をサポートします。
SC22 における NVIDIA の取り組みを知ると共に、特別講演のリプレイもご覧ください。
※ブログトップの画像は、本研究チームの開発したLLMによって配列決定された新型コロナ株。各ドットは変異株によって色分けされています。画像提供:アルゴンヌ国立研究所のBharat Kale氏、Max Zvyagin氏、Michael E. Papka氏